

继上期《美国原料药/辅料/包材DMF备案登记全流程解析》系统梳理了DMF备案的五大类型及费用结构后,本期将重点剖析DMF备案全流程,涵盖提交方式、登记步骤和审评要点等关键环节,旨在助力企业精准规划、高效备案,顺利进入美国市场。
DMF资料提交及登记流程
01 提交方式
自2017年5月起,所有的新药申请(NDA)、仿制药申请(ANDA)、生物制品申请(BLA)及其补充申请必须强制以e-CTD格式提交;自2018年5月起,FDA要求商业化IND和药物主文件(DMF,III类DMF除外)申报资料必须以e-CTD格式提交,其他所有格式将被拒绝。
应用e-CTD的申报流程
①CTD资料撰写:CTD一般由 5 个模块组成,每个模块包含各类药品注册资料的要求。
②CTD资料文档编辑:在准备好电子版材料后,需将其转化为美国FDA接受的电子文档形式。
③电子资料提交:目前美国以2种途径进行提交:一是以电子提交通道(Electronic Submission Gateway, ESG)提交,二是使用物理媒介(例如光盘CD-ROM)。对于文件大小在10GB以下的注册资料(大部分DMF都不超过10GB),均可通过ESG提交。若文件大小超过10GB,则需通过光盘提交。
02 DMF登记流程
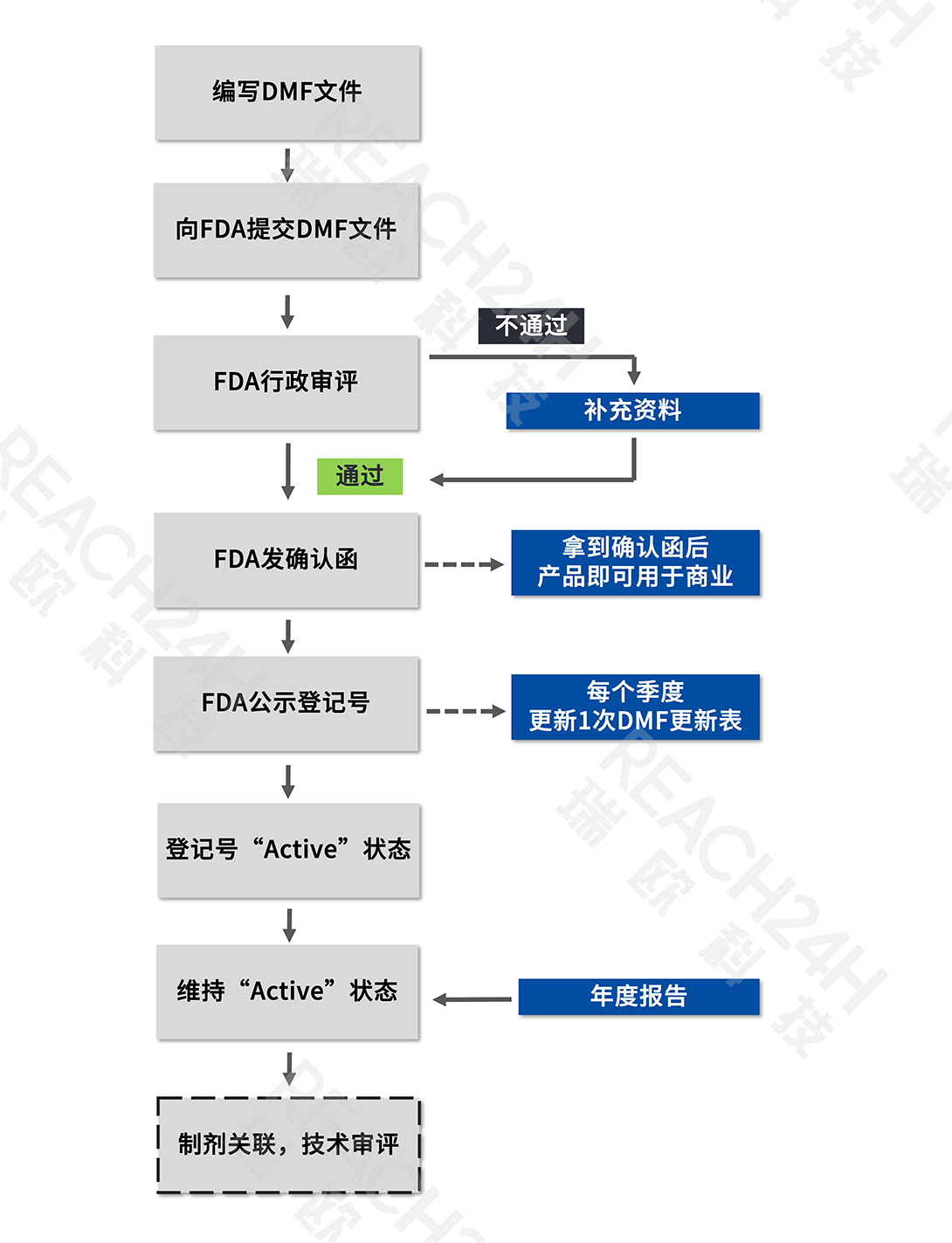
Inactive:未激活状态,意味着DMF被DMF持有人或FDA关闭。
03 DMF审评流程
①提交DMF文件前,需先申请得到DMF预分配号;
②资料接收后,FDA将对原始DMF文件进行行政审评,审核资料形式完整性。
③行政审评预计花费2-3周(注:具体时间视FDA的客观情况而定,下同)。
④行政审评结果有以下2种情形:
情形1:审评通过
FDA将向DMF持有人发送确认函(Acknowledgement Letter),列入DMF清单。该DMF的状态转为“激活/A”。
情形2:审评未通过
FDA将通知DMF持有人存在的缺陷项。修正缺陷后,DMF的状态才能转为“激活/A”
⑤确认函包括以下关键信息:DMF登记号,提交日期,DMF类型、主题(标题)、持有人、提交方和代理人等。

确认函示例
①对于Ⅱ类API,完成缴费后,FDA开始完整性审评(CA)。
②审评合格将被列入CA清单。审评不合格,申报方会收到FDA发布的缺陷信,并要求在限定期限内完成回复。
③完整性审评预计花费60天。
技术审评
①Ⅱ类API被ANDA引用后,FDA会启动技术审评,用于支持其特定用途。
②DMF持有人可能会因为制剂产品的初次上报、变更等收到相应缺陷信,无缺陷后,则会颁发首次完善信。
③技术审评时间基于制剂审评周期而定。
DMF备案登记常见问题
Q1:DMF登记是否一定需要美国代理?
A:为促进沟通,FDA强烈建议非美国本土DMF持有人指定一位熟悉FDA法规、指南和程序的美国代理人,代理人可以代表DMF持有人向FDA提交文件。
Q2:何种情况会导致DMF关闭?
A:DMF的关闭可能是因为DMF持有人要求关闭,也可能是因为FDA无法确定该DMF是否有效。在后一种情况下,FDA将通知持有人或代理人,DMF需要更新。如果DMF持有人或代理人未及时提交年度报告,FDA可能会关闭该DMF。
Q3:什么情况下需要补充资料?
Q4:DMF资料提交后多久可以获得登记号?
A:在提交e-CTD格式的DMF文件前,DMF持有人需先向CDER/CBER 申请预分配号(Pre-Assigned Application Number),获得该号后再提交DMF文件。随后,FDA将启动行政审评,审评通过后,FDA会发送确认函,其中包括DMF登记号。
Q5:如何查询DMF登记号?
A:通过行政审评并收到确认函后,并不能立即在FDA官网查询到该登记号。FDA每季度更新一次DMF列表,需待下一次更新后,才会显示该DMF号的“A”状态。
Q6:之前提交的DMF文件是否需要转化为e-CTD格式?
A:FDA并未强制要求将之前已提交的DMF转化为e-CTD格式。如DMF持有人自愿转化,可通过电子提交通道(ESG)重新提交转化后的e-CTD格式文件。是否转化格式不影响后期以e-CTD格式提交年报或变更。
相关阅读:
超详细!美国原料药/辅料/包材DMF备案登记全流程解析(含实操要点)




 化学品合规
化学品合规
 化妆品合规
化妆品合规
 检验检测
检验检测
 安全管理智能化
安全管理智能化
 绿色低碳可持续
绿色低碳可持续
 食品合规
食品合规
 食品接触材料/再生塑料
食品接触材料/再生塑料
 中国农药登记
中国农药登记
 境外农药登记
境外农药登记